

Lysozyme was pivotal in establishing how a protein's 3D shape determines its function, enhancing our understanding of protein structure-function relationships.
Whereas the elegant cube-like structure of salt (NaCl) was met with what was essentially disbelieving anger, Francis Crick famously asked at the 1965 public presentation of Lysozyme’s structure, “Could that be where the substrate polysaccharide binds?”. He was pointing at the mouth-like cleft of the enzyme, which did turn out to be precisely the active site; the jaws with which Lysozyme bites at the protective peptidoglycan coat of gram-positive bacteria. Since then, Lysozyme has found itself used widely as a model protein due to its small size, stability, and availability in large quantities. Lysozyme is commonly used as the model protein to benchmark new technological developments in X-ray crystallography, crystallization, and a host of others. It is often the first protein used in teaching X-ray crystallography to students, a process that today takes minutes rather than months.
Solving the Structure today
Today’s technological developments mean that computational solving of protein structures is highly advanced, to the extent that the most complex and time-consuming part of the process is getting the protein to crystallize. This computational (in-silico) structure-solving, or protein folding, recently hit headline news globally with the announcement of AlphaFold. Developed by Google-owned DeepMind, AlphaFold is an artificial intelligence system that predicts protein structures entirely from the amino acid sequence. This is achieved using the “evolutionary, physical, and geometric constraints” of protein structures. For those inclined, the open-access announcement paper can be found here:
J Jumper et al. 2021 Highly accurate protein structure prediction with AlphaFold
Solving protein structures using just the amino acid sequence isn’t a new idea, however. The possibility of such an approach was first suggested over 60 years ago.
___
“It seems a reasonable assumption that, as the synthesis proceeds, the amino end of the chain becomes separated by an increasing distance from the point of attachment to the ribosome and that the folding of the protein chain to its native conformation begins at this end even before the synthesis is complete. According to our present ideas, parts of the polypeptide chain, particularly those near the terminal amino end, may fold into stable conformations that can still be recognized in the finished molecule and that act as 'internal templates,' or centers, around which the rest of the chain is folded.“
Sir David Phillips 1966, Scientific American
___
As a protein is produced, the information is nearly entirely inherent in the amino acid sequence that emerges like a string of beads from its constructor, the ribosome. Each amino acid has its own properties, some being attracted to water (hydrophilic), others entirely repelled (hydrophobic), and some carrying a positive charge with negatively charged neighbors. As these beads emerge one after the other, the protein begins to take the shape which works best for its string of members, shielding the water-repelling amino acids on the inside while exposing water-loving amino acids to the outer surface. In this way, amino acids that are not direct neighbors in this chain of beads can become closely associated as they begin to bend and fold into their 3D shape. The functional shapes that are created endow the protein, in our case, an enzyme, with its abilities. For Lysozyme, it is clear that its folding gives rise to a prominent central channel, a void within which the peptidoglycan of gram-positive bacteria meets its demise. For AlphaFold, the information with which to predict the 3D structure of proteins is entirely within this amino acid chain
___
“The properties our networks predict are: (a) the distances between pairs of amino acids and (b) the angles between chemical bonds that connect those amino acids. The first development is an advance on commonly used techniques that estimate whether pairs of amino acids are near each other.”
Google DeepMind, January 2020
___
Our own model
Unlike the lab of Sir. Lawrence Bragg, we do not need linear diffractometers, nor X-Ray generators to render our own model of Lysozyme. Neither do we need a Ferranti Mercury, the early computer that used 2000 vacuum tubes, occupied an entire room, weighed 1100 kg, and cost the equivalent of $3,800,000. For us today it is simpler, much of the laboratory equipment required for the trial-and-error process of crystallising proteins can be automated. Interpretation and measuring of diffraction images are now in-silico, and amino acid sequences of a huge number of proteins are easy to find if you know where to look. But, it all started somewhere. And that is with the very first proteins to be crystallised, which gave revelations as to how the shape of a protein defines its function Making our own 3D model is a simple matter of finding the protein’s unique identifying code (in our case: 1DPX), accessing the publicly available dataset on PDB (protein data bank, see here and passing that into free software from UCSF, Chimera. A little fiddle with the settings makes our structures a little more amenable to 3D printing, and we can send the generated file off to those that know better for the end product to be sprung into life, layer by deposited plastic layer. As I sit here writing this, the test 3D print of Lysozyme looks back at me from my desk. It’s hard to ignore its deep central cleft and to think of the growing excitement that the team must have shared when first building their labor- and intellect-intensive model, sheet by sheet, to reveal this elegant little enzyme.
3D Model 1: Lysozyme - Volume Model version
Click & Drag to explore | Use the mouse wheel to zoom
This experiment is built for Google Chrome and won't work properly on other browsers such as Firefox and Edge.
Click & Drag to explore | Use the mouse wheel to zoom
This experiment is built for Google Chrome and won't work properly on other browsers such as Firefox and Edge.
3D Model 3: Lysozyme - Licorice Model version
Click & Drag to explore | Use the mouse wheel to zoom
This experiment is built for Google Chrome and won't work properly on other browsers such as Firefox and Edge.
How does DeepMind’s AlphaFold perform?
The structure we used as the ground truth to print our model was the result of painstaking X-ray crystallography work. This required significant man-hours and culminated in a publication (Weiss MS et al. Acta Crystallographica Section D 2000). AlphaFold needs no experimentation; however, only the amino acid sequence found here:
UniProtKB - P00698 (LYSC_CHICK).
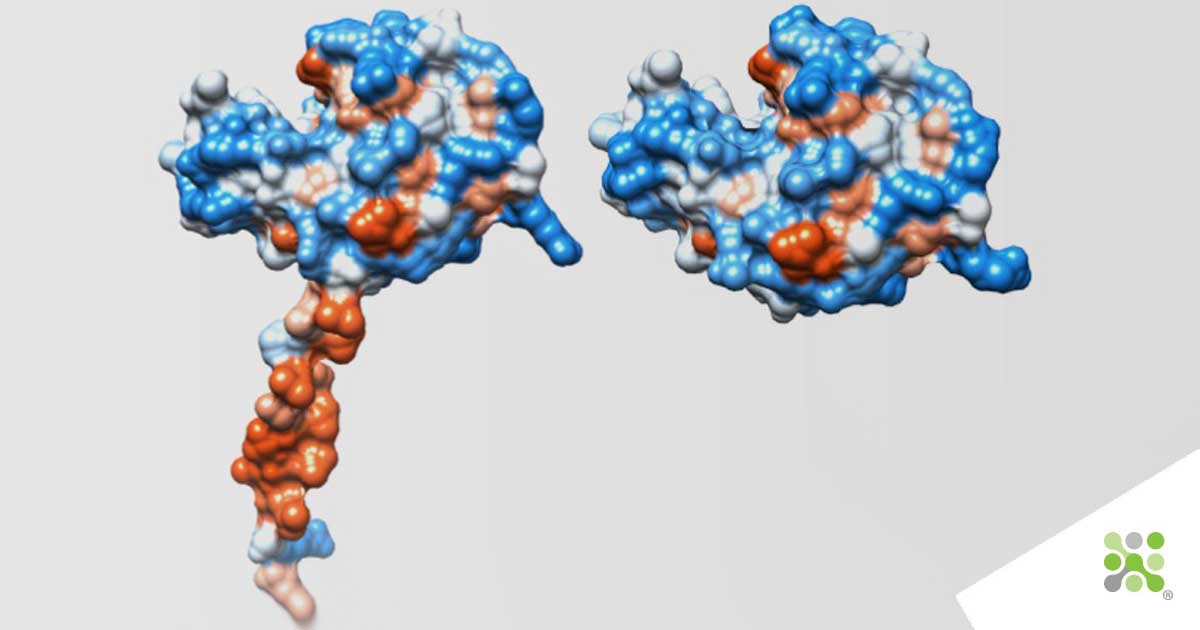
Fig. 1: Left: Structure predicted by DeepMind AlphaFold AI for Chicken Lysozyme.
Right: X-ray crystallographic structure of Chicken Lysozyme as solved in 2000, with 1.65 Angstrom resolution

Fig. 2: Fig. 2: Particle Size Analysis by X-Ray Diffraction

Fig. 3: A Ferranti Mercury computer
We looked at the AlphaFold database to examine their predicted structure for chicken Lysozyme. Interestingly, AlphaFold performs extremely well, for the most part, resolving the catalytic cleft of Lysozyme with visual similarity, see the image above. What might strike you is the handle-like protrusion at the bottom of the model, “surely that’s rubbish”, you might be saying. Well, you can keep that linear diffractometer and X-ray generator stored away for now. I thought the same until I dug a little deeper into the data used by DeepMind. For their model, they have actually used an amino-acid sequence that includes the signal sequence of Lysozyme. This signal sequence is the processing limb of the freshly made protein, used to signal to the cell’s transport machinery to literally grab it, and shuttle it outside of the cell. As Lysozyme’s functionality is needed on the outside of the cell, it needs to signal with this component that it needs to be released/secreted, at which point the limb is shed. So… the handle is indeed a handle! The question that remains for me is What would the Braggs make of all of this?
Some video resources for the keen
In our research, for this installment, we have come across a beautiful selection of videos produced by the Royal Institution and others about Lysozyme, X-ray crystallography, and similar. We thought it would be valuable to share these with anybody interested in delving a little deeper into these topics.
The humble Braggs and X-ray crystallography: Solving the patterns of matter
A video showing the crystallisation of Lysozyme
DeepMind’s own video on protein crystallography
100 years of Lysozyme
2022 marks 100 years since news of Lysozyme’s discovery reached the Royal Society. Let us take you on the journey of this 100-year story:
Lysozyme leads the way for DeepMind’s AlphaFold
Lysozyme contributed directly to our understanding of the structure-function relationship of proteins. It was the first solved structure that established the concept that the 3D shape of a protein[...]

A beginning for Lysozyme
Bioseutica® group has been producing lysozyme from hen egg-white since the 1950s, a history spanning nearly 75 years. But lysozyme’s story doesn’t begin with us. For that, we must look back to the earliest human use of egg-white[...]

Thus ended the collection of tears
The second installment of our 100 Years of Lysozyme series is now live. This week, we discover how the abundance of Lysozyme in eggs was first discovered (hint: it wasn’t in the supermarket)[...]

Not a great lecturer
Fleming’s shyness, his being “not a great lecturer” as acknowledged by Allison, may in part account for the poor reception received at the first reading of the initial paper describing lysozyme[...]

Try £ 3
From 3p for tears to £3 for Picassos; in this installment, we uncover how Alexander Fleming’s eye and contributions had an immeasurable impact on medicine and science in the 20th century[...]

A Tradition of Innovation
The aftermath of the war has left Italy economically devastated like the rest of the European countries. In Milan, another Ferrari begins production, but this time of Penicillin.[...]

A Tradition of Innovation 2
The death of their first trial patient through the insufficient supply of Penicillin drove Florey, Chain, and their research assistant, Heatley, forward with inspired momentum.[...]

A Tradition of Innovation 3
24th May 1947 saw the establishment of SPA (Società Prodotti Antibiotici) by Pharmacologist Dr. Rodolfo Ferrari and microbiologist Carlo Callerio. Now Italy had its own, domestic Penicillin[...]

More than an antimicrobial
To most of us, if at least those reading, I hope, Lysozyme is best known as a potent, natural antimicrobial. This is evidently its primary purpose for the hen's egg, making[...]

Finding the structure
Sir. William Bragg and Sir. Lawrence Bragg shared the Nobel Prize in 1915 for their work on analyzing crystal structures using X-Rays. This award is notable not only as it was the first and [...]